境界哥网赚|引流|灰产|揭秘|思维|经验|关注
越是长尾流量,用户的需求越精准越是精准需求,越容易形成暴利产品这句话,不是我说的,而是某产品专家,总结了十来年的经验说出了这句话,我认为很经典分享给大家既然是这样的,那么我们如何发现一些精准市场,并且竞争不是太大的行业呢?今天我就教大家一个方法,快速去发掘此类蓝海市场。
1. 关键词发掘长尾关键词主关键词大家都知道,一般适用于行业词。但是我们今天讲的这个主关键词,就是口语词,疑问词,这类词里面包含着很多非常不错的暴利行业。
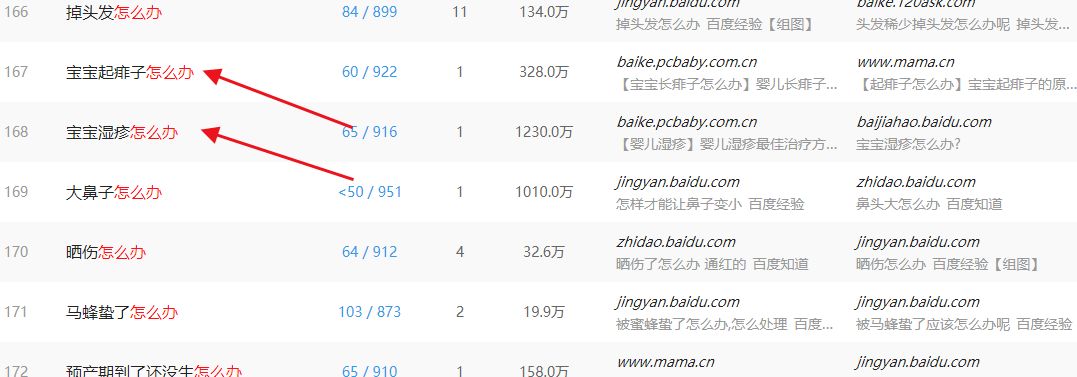
我们用疑问词搜索,大家会发现,网民们有许多各种各样的状况,不知道该如何解决就会用 怎么办 来问大家可以看到,一般的口语词 疑问词 ,到一百八十几位的时候,基本上流量已经很少了而这个怎么办,排到现在 竟然还有那么高的流量。
只能证明一个问题:寻求解决办法的人太多了2. 筛选词筛选词,是数据分析的一项基本功,其宗旨就是从一堆数据里面筛选出有用的来对于我们分析市场行业来讲,这个时候的筛选,我们要做的就是筛选出竞争大的,去除,筛选出利润小的去除,筛选出资源具有不可复制性的去除,筛选出没有商业价值的去除。
留下的就是竞争小,操作难度小,资源具有复制性的。经过我的一番筛选,找出一个竞争难度小的,并且暴利的产业。
我们看到关于宝宝怎么办中的两个关键词搜索量都是不错的。我们具体看一下搜索数据。
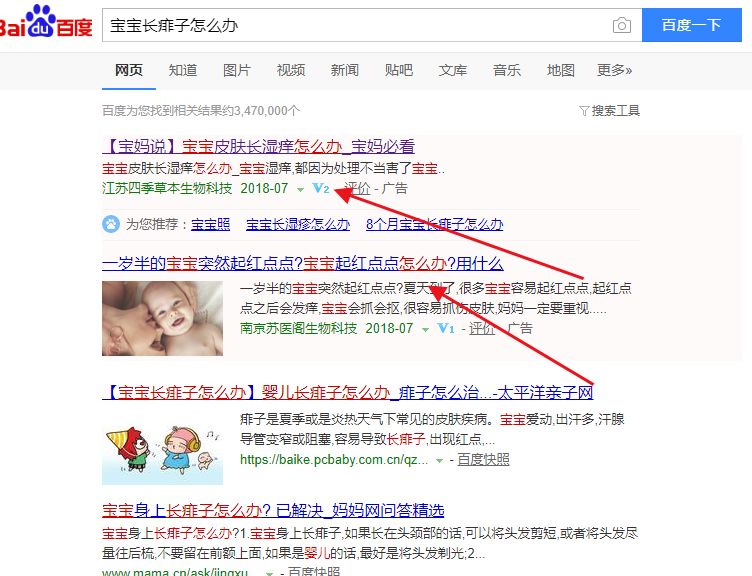
这个时候,我们能够看到上面有两个竞价广告而且第一个一看就知道投放广告时间比较长了,因为已经到了V2级还有竞价下面排名最高的竟然是太平洋亲子网,说明优化难度并不高讲到这里,我跟大家说一点小诀窍:我们在发现这些暴利行业的时候,最容易忽略一个问题就是,我们分析关键词有搜索量,但是打开搜索却没有竞价。
这个时候不要高兴的太早,因为造成这种情况的有三种原因:1.有些关键词本身没有商业价值,客户成交率低下,收不回本,也不投放了2. 分时间段投放,意向客户搜索的时间段集中在一个点上,所以到点才投放3.第三种就是蓝海市场。
不过现在已经不存在完全蓝海的市场了,毕竟从业人群多了,很多都是有竞争的我们能做的就是发现一些竞争比较小的,我们进去分杯羹好了,确定了竞争不是很激烈的时候,我们要开始下一步动作。3. 搜集相关关键词

这个时候,我们能够看到,起痱子,湿疹,红屁股,还有长痱子。都是相关关键词。4.根据关键词寻找产品
经过价格对比,找到了这款售价48元的,儿肤宝而且评价效果不错,这个时候,有两种方案去做,要卖高价,就换包装一般价格就不用换了竞价一些产品本身进价贵,效果好,为追求复购率以及高利润,这类方法常用如果换包装的话,直接淘宝搜包装盒就行了。
很多号称老中医的产品都是这么来的
只有你想不到的,只要你能想到的,大部分都有卖的搞定了产品,继续下一步5. 寻找素材如果你经常观察竞价广告的话,你会发现他们都通过引导到微信上来成交我是特别喜欢微信成交,比较便捷,也有支付场景一旦加上,就能够长期营销。
就像这样的我们让人加了微信之后,朋友圈总的有内容,要不然很多人不能及时通过,有人就会忘记加你的目的其实素材寻找有两种,一种是同行,另外一种还是同行只不做平台不一样,一种是竞价同行,一种是淘宝同行素材特别多,毕竟专业做这个的。

就像这样触目惊心的素材,很多的做产品做的多,你朋友圈发的图片多了,你会明白一个道理:越是反差大的图片,越能够激发顾客购买欲望6. 开始推广前面五项是项目基础,最重要的环节来了推广环节,该如何推广其实我是不建议新手盲目的做付费广告的,因为没个几千上万块钱,很难摸索出门道来。
因为你对互联网推广的本质没认识清楚玩推广,如果你免费层的推广玩的很好,付费照样牛逼这是循序渐进的过程,因为免费玩的好的话,你对顾客越了解,对关键词精准度越能拿捏的准免费推广究竟如何做?把我们刚才整理出来的关键词,这个时候派上用场了。
我们首先要做的就是关键词标题组合
百度搜索爆文标题生成器,就能找到很多类似的软件组合好爆文标题之后,我们开始推广1. 百度贴吧百度贴吧答疑贴首先确定范围,我们解决宝宝的问题,客户自然是宝妈在宝妈吧发帖就是最好的选择,当然想找更多的贴吧自然是用贴吧推荐。
就像这样,会自动给你推送很多贴吧帖子标题取名一定要一针见血,划定范围,比如:我是一名儿童科皮肤医生,有什么问题,可以留言,免费答疑 类似这样的标题,往往更能获取关注当然,发广告,要用楼层回复,效果更好2.
百家号在百家号发文章的原因是易收录,而且排名不错整合关键词相关文章,发到百家号里,不允许留联系方式就在图片打水印3. 天涯天涯发同名帖子,也能获得不错的排名4. 博客开通博客的原因是能短时间内获得排名,以及增加权威性。
5. 头条头条屌丝多,这是不争的事实,但是宝妈群体付费率高,也是事实写相关文章,普及然后评论区,用小号提问,大号回复联系方式把这五个推广渠道玩好了,每天吸引个30-50个精准粉丝,不在话下,而且成交率特别高。
总结:完整操盘一个项目,你会对互联网有了更加深刻的认知,对于各类项目操作更加有深刻的体验如果有对此文有任何看法和观点,欢迎留言讨论如果你想了解哪些热门项目,也可以下方留言,说不准下一个分析的项目就是你提出来的。
要做网赚项目的朋友可以在公众号菜单领取,境界哥本人操作的项目都有。实时更新。 识别下方二维码,与境界哥微信交流。
往期精彩回顾关于我和公众号互联网逆袭必看,速度把握,机会难得……让小白在互联网上逆袭赚大钱的机会,机会很少,请珍惜……境界哥主要分享网赚、自媒体类的干货,项目因人而异,大家可以选择适合自己的项目,文章如有不对之处,欢迎前来交流,指正!
点击 阅读原文 看社群动态
亲爱的读者们,感谢您花时间阅读本文。如果您对本文有任何疑问或建议,请随时联系我。我非常乐意与您交流。






发表评论:
◎欢迎参与讨论,请在这里发表您的看法、交流您的观点。